多基因组比对
简介
多基因组比对分为有参考基因组(Referenced alignment)和无参考基因组(Reference-free alignment)两种情况,区别是有参考基因组的比对只会报告包含参考基因组的比对,忽略其他基因组之间的比对。MULTIZ属于有参考基因组的比对。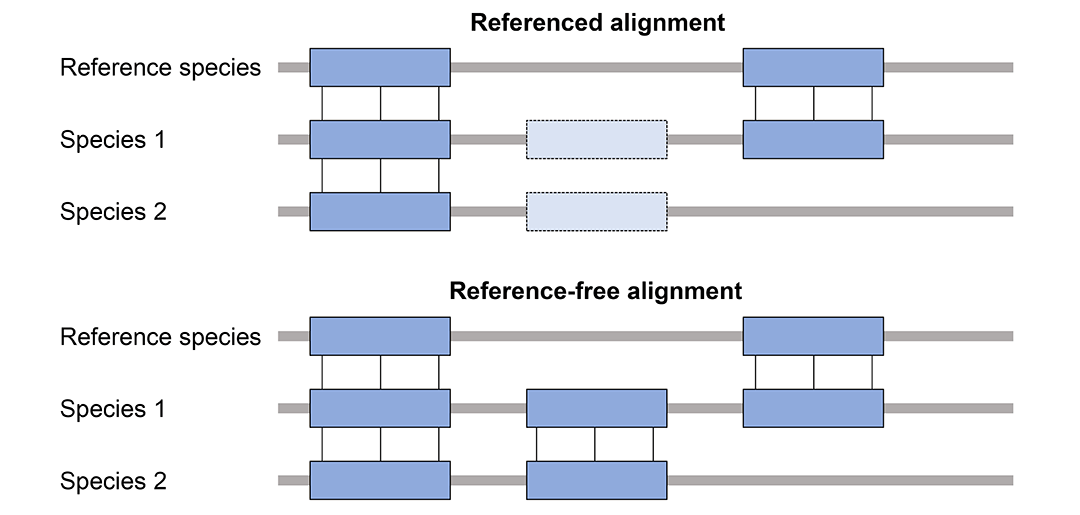
MULTIZ这套方法也被称作TBA(threaded blockset aligner)或者MULTIZ / TBA。MULTIZ与LASTZ不同,它不是一个真的序列比对软件。它利用基因组两两比对的结果,结合它们的进化树,得到多基因组比对结果。比如,参考物种ref_species和species_1、species_2的多基因组比对,先分别做ref_species和species_1比对、ref_species和species_2比对,然后用两两比对的结果和ref_species、species_1、species_2的进化树作为输入,利用MULTIZ得到多基因组比对结果。
安装MULTIZ
同LASTZ一样,MULTZ也不再更新了,我们使用Github上的可以被现代编译器编译的版本。
1 | cd ~/software/ |
运行MULTIZ
我们这里以斑马鱼(Danio rerio)、鲤鱼(Cyprinus carpio)、鲫鱼(Carassius auratus)基因组为例,斑马鱼的基因组作为参考基因组。假设我们已经通过两基因组比对,得到了Danio_rerio.Cyprinus_carpio.maf和Danio_rerio.Carassius_auratus.maf。
1 | ln -s Danio_rerio.Cyprinus_carpio Danio_rerio.Cyprinus_carpio.sing.maf |
输入的MAF文件的命名模式为ref_species.species_1.sing.maf,进化树的格式为关于Newick格式(Newick tree format)。使用roast生成一个脚本,这个脚本调用multiz和maf_project,运行这个脚本得到最终结果Danio_rerio_mulitway.maf。