Introduction
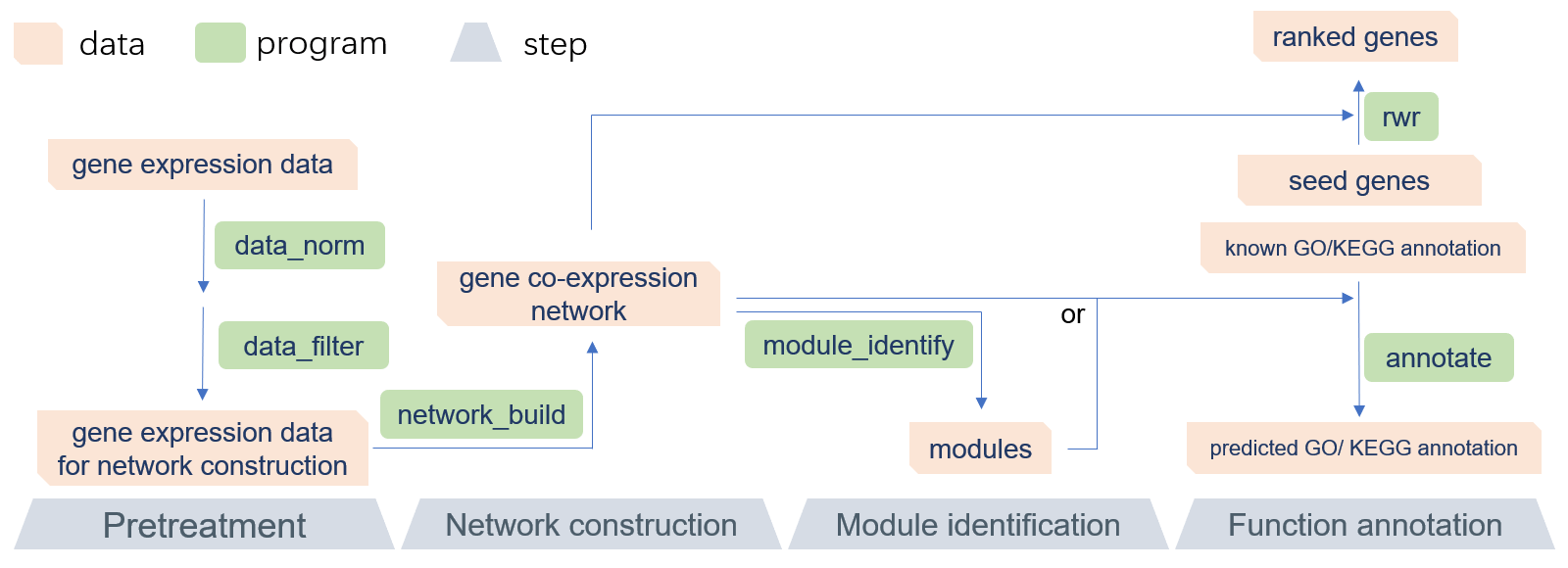
GCEN workflow
GCEN is a command-line toolkit that allows biologists to easily build gene co-expression network and predict gene function, especially in RNA-Seq research or lncRNAs annotation. GCEN is primarily designed to be used in lncRNAs annotation, but is not limited to those scenarios. It is an efficient and easy-to-use solution that will allow everyone to perform gene co-expression network analysis without sophisticated programming skills. The recommended pipeline consists of four parts: data pretreatment, network construction, module identification, and function annotation. A README file and sample data are included in the software package. Because of its modular design, the GCEN can be easily integrated into another pipeline. Also, the multithreaded implementation of GCEN makes it fast and efficient for RNA-Seq data.
Usage
Main programs
description: The program data_norm normalizes the gene expression data. usage: data_norm -i input_file -o output_file -m normalization_method options: -i --input <input file> -o --output <output file> -m --method <upqt | median | deseq | tmm | hkg> normalization method (default: upqt) -g --gene <housekeeping gene list> only for '--method hkg' -v --version display GCEN version -h --help print help information example: data_norm -i ../sample_data/gene_expr.tsv -o ../sample_data/gene_expr_norm.tsv -m tmm
description: The program data_filter filter genes according to the their expression mean and standard deviation. usage: data_filter -i input_file -o output_file options: -i --input <input file> -o --output <output file> -c --cutoff_mean <number> mean cutoff of gene expression (default: 0.0) -C --cutoff_sd <number> standard deviation cutoff of gene expression (default: 0.0) -p --percent_mean <number> keep a proportion of total genes based mean of gene expression (default: 1.0) -P --percent_sd <number> keep a proportion of total genes based standard deviation of gene expression (default: 1.0) -v --version display GCEN version -h --help print help information example: data_filter -i ../sample_data/gene_expr.tsv -o ../sample_data/gene_expr_filter.tsv -p 0.75
description: The program network_build construct gene co-expression network using gene expression matrix. usage: network_build -i gene_expr.tsv -o gene_co_expr.network options: -i --input <input file> -o --output <output file> -m --method <pearson or spearman> correlation coefficient method (default: spearman) -l --log <log | log2 | log10> make a log(x+1) transformation (default: not transform) -t --thread <number> cpu cores (default: 2) -p --pval <number> p value cutoff (default: 0.001) -c --cor <number> correlation coefficient cutoff (default: 0.1) -s --signed <y or n> singed network (default: n) -f --fdr <y or n> calculate FDR (default: n) -a --all output all edges without any cutoff (if -a is specified, the -p and -c are ignored) -v --version display GCEN version -h --help print help information example: network_build -i ../sample_data/gene_expr.tsv -o ../sample_data/gene_co_expr.network -m spearman -p 0.001 -f
description: The program identify the gene modules using the gene co-expression network. usage: module_identify -i input_file -o output_file options: -i --input <input file> -o --output <output file> -s --similarity <number> similarity cutoff (default: 0.5) -t --thread <number> cpu cores (default: 2) -v --version display GCEN version -h --help print help information example: module_identify -i ../sample_data/gene_co_expr.network -o ../sample_data/module.txt
description: The program annotate can perform GO/KEGG annotation based on network or module. usage: annotate -g go-basic.obo -a gene_go_association_file -n input_network -o out_dir options: -g --go <go-basic.obo file> -k --kegg <kegg information> (if the -g/--go is specified, the -k/--kegg are ignored) -a --assoc <gene-GO/KEGG association file> -n --network <network file> -m --module <module file> (if -n is specified, the -m is ignored) -p --pval <number> p value cutoff (default: 0.05) -o --output <output directory> -t --thread <number> cpu cores (default: 2) -v --version display GCEN version -h --help print help information examples: ./annotate -g ../sample_data/go-basic.obo -a ../sample_data/gene_go.assoc -n ../sample_data/gene_co_expr.network -o ../sample_data/network_go_annotation ./annotate -g ../sample_data/go-basic.obo -a ../sample_data/gene_go.assoc -m ../sample_data/module.txt -o ../sample_data/module_go_annotation ./annotate -k ../sample_data/K2ko.tsv -a ../sample_data/gene_kegg.assoc -n ../sample_data/gene_co_expr.network -o ../sample_data/network_kegg_annotation ./annotate -k ../sample_data/K2ko.tsv -a ../sample_data/gene_kegg.assoc -m ./sample_data/module.txt -o ../sample_data/module_kegg_annotation
description: The program rwr can predict potential funcation associated genes based on RWR (Random Walk with Restart) algorithm. usage: rwr -n input_network -g gene_list -o output_result options: -n --network <network file> -g --gene <seed genes list file> -r --gamma <number> restart probability (default: 0.5) -p --prob <number> probability cutoff (defalut: 0.01) -o --output <output file> -d --directed_network the input network is directed (defalut: the input network is undirected) -w --weighted_network the edge weights of network will be considered (defalut: all edge weights of network are set to 1.0) -W --weighted_gene the weights of seed genes will be considered (defalut: all weights of seed genes are set to 1.0) -v --version display GCEN version -h --help print help information example: rwr -n ../sample_data/gene_co_expr.network -g ../sample_data/rwr_seed_genes.list -o ../sample_data/rwr_ranked_gene.tsv
Utilities
description: The program csv_to_tsv convert CSV (Comma-Separated Values) file to TSV (Tab-Separated Values) file. usage: csv_to_tsv -i input.csv -o output.tsv options: -i --input <input csv file> -o --output <output tsv file> -v --version display GCEN version -h --help print help information example: csv_to_tsv -i ../sample_data/gene_expr.csv -o ../sample_data/gene_expr.tsv
description: The program tsv_to_csv convert file TSV (Tab-Separated Values) to CSV (Comma-Separated Values) file. usage: tsv_to_csv -i input.tsv -o output.csv options: -i --input <input tsv file> -o --output <output csv file> -v --version display GCEN version -h --help print help information example: csv_to_tsv -i ../sample_data/gene_expr.tsv -o ../sample_data/gene_expr.csv
description: The program data_stat calculate the statistics of gene expression matrix. usage: data_stat -i input_file options: -i --input <input file> -v --version display GCEN version -h --help print help information example: data_stat -i ../sample_data/gene_expr.tsv
description: The program network_stat calculates the statistics of network. usage: network_stat -i input_file options: -i --input <input file> -v --version display GCEN version -h --help print help information example: network_stat -i ../sample_data/gene_co_expr.network
description: The program network_merge merge two or more networks. usage: network_merge -i input_files -o output_file options: -i --input <input files> multiple files are separated by commas -o --output <output file> -c --cor <number> correlation coefficient cutoff (default: 0.5) -h --help print help information example: network_merge -i ../sample_data/test_1.network,../sample_data/test_2.network -o ../sample_data/test_merge.network
description: The program network_extract extracts subnetwork based on a gene list. usage: network_extract -i input.network -g gene_list.txt -o output.network options: -i --input <input network file> -o --output <output network file> -g --gene <gene list file> -v --version display GCEN version -h --help print help information example: network_extract -i ../sample_data/gene_co_expr.network -g ../sample_data/gene_list.txt -o ../sample_data/sub.network
description: The program network_shuffle random shuffles network with degree preserving. usage: network_shuffle -i input.network -o output.network options: -i --input <input network file> -o --output <output network file> -s --swap <number> multiples of edges number for double-edge swaps to perform (default 100) -v --version display GCEN version -h --help print help information example: network_shuffle -i ../sample_data/test_1.network -o ../sample_data/random_shuffled.network
description: The program calculate_accuracy calculate the accuracy of prediction. usage: calculate_accuracy -g go-basic.obo -a gene_go_association_file -n input_network options: -g --go <go-basic.obo file> -a --assoc <gene-GO/KEGG association file> -n --network <network file> -p --pval <number> p value cutoff (default: 0.05) -t --thread <number> cpu cores (default: 2) -v --version display GCEN version -h --help print help information examples: ./calculate_accuracy -g ../sample_data/go-basic.obo -a ../sample_data/gene_go.assoc -n ../sample_data/gene_co_expr.network
description:
The program enrich can perform GO/KEGG enrichment.
usage:
enrich -e enrichment_gene_list_file -b background_gene_list_file -g go-basic.obo -a gene_go_association_file -p p_value_cutoff -o out_put_file
options:
-e --enrich <enrichment gene list file>
-b --background <background gene list file>
-g --go <go-basic.obo file>
-k --kegg <kegg information> (if the -g/--go is specified, the -k/--kegg are ignored)
-a --assoc <gene/go association file>
-p --pval <number> p value cutoff (default: 0.05)
-o --output <output file>
-v --version display GCEN version
-h --help print help information
examples:
enrich -e ../sample_data/enrichment_gene.list -b ../sample_data/background_gene.list -g ../sample_data/go-basic.obo -a ../sample_data/gene_go.assoc -p 0.05 -o ../sample_data/enrichment.go
enrich -e ../sample_data/enrichment_gene.list -b ../sample_data/background_gene.list -k ../sample_data/K2ko.tsv -a ../sample_data/gene_kegg.assoc -p 0.05 -o ../sample_data/enrichment.kegg
generate_expr_matrix_from_rsem
description: The program generate_expr_matrix_from_rsem generate gene expression matrix from RSEM outputs. usage: generate_expr_matrix_from_rsem -i input_file -o output_file options: -i --input <input file> a text file with sample ID and path to its RSEM result file on each line -o --output <output file> -t --tpm output TPM value instead of FPKM vaule -v --version display GCEN version -h --help print help information example: generate_expr_matrix_from_rsem -i ../sample_data/rsem/rsem_sample.txt -o ../sample_data/rsem/rsem_gene_expr.tsv
generate_expr_matrix_from_stringtie
description: The program generate_expr_matrix_from_stringtie generate gene expression matrix from StringTie outputs. usage: generate_expr_matrix_from_stringtie -i input_file -o output_file options: -i --input <input file> a text file with sample ID and path to its GTF file on each line -o --output <output file> -t --tpm output TMP value instead of FPKM vaule -v --version display GCEN version -h --help print help information example: generate_expr_matrix_from_stringtie -i ../sample_data/stringtie/stringtie_sample.txt -o ../sample_data/stringtie/stringtie_gene_expr.tsv
Data format
To understand the format of the input and output files for each program, please take a look at the sample data included in the software package.
Most input and output files of GCEN are tab-separated values (TSV) files. We provide two programs csv_to_tsv and tsv_to_csv for converting TSV and CSV files to each other.
Gene expression matrix
The first column are gene name, the others are gene expression value.
#gene SRR372787 SRR372788 SRR372789 SRR372790 SRR372791 SRR372792 ENSDARG00000117464 0.00 55.64 8.33 219.56 0.00 249.22 ENSDARG00000117133 0.00 21.20 0.00 18.15 0.00 29.70 ENSDARG00000117089 62.73 55.50 318.04 240.76 96.14 69.75 ENSDARG00000116743 18.43 219.98 6.71 189.01 11.30 221.65 ENSDARG00000115987 0.00 92.30 0.00 78.53 24.40 108.66
Gene co-expressiong network
Each column is gene a, gene b, correlation coefficent, p-value, FDR.
#node1 node2 correlation p-value FDR ENSDARG00000039935 ENSDARG00000062108 0.881139 2.988639e-06 8.097798e-05 ENSDARG00000039935 ENSDARG00000016088 0.867002 6.652075e-06 1.420293e-04 ENSDARG00000039935 ENSDARG00000099024 0.823607 4.842646e-05 5.762282e-04 ENSDARG00000039935 ENSDARG00000001803 0.805322 9.588506e-05 9.389785e-04 ENSDARG00000039935 ENSDARG00000019835 0.885008 2.359077e-06 6.912051e-05
Known gene annotation
The first column are genename, the others are annotation. For genes with multiple annotations, a format with multiple rows (two columns per row, for gene and annotation) per gene is also acceptable.
ENSG00000211780 GO:0005886 ENSG00000211780 GO:0002250 ENSG00000211664 GO:0002377 GO:0005886 GO:0006955 GO:0005615 GO:0003823 GO:0002250 ENSG00000231292 GO:0002377 GO:0005615 GO:0006955 ENSG00000271383 GO:0005737
Predicted gene GO annotation
GO name name_space enrichment study_count study_n pop_count pop_n p_val GO:0035672 oligopeptide transmembrane transport biological_process e 3 49 3 973 0.000120375 GO:0006857 oligopeptide transport biological_process e 3 49 3 973 0.000120375 GO:0044281 small molecule metabolic process biological_process e 12 49 91 973 0.00109452 GO:0006766 vitamin metabolic process biological_process e 3 49 5 973 0.00111971 GO:0006446 regulation of translational initiation biological_process e 3 49 5 973 0.00111971
Predicted gene KEGG annotation
ko name enrichment study_count study_n pop_count pop_n p_val ko02010 ABC transporters e 4 50 7 443 0.00391765 ko04070 Phosphatidylinositol signaling system e 2 50 3 443 0.0348134 ko04011 MAPK signaling pathway - yeast e 2 50 3 443 0.0348134 ko04977 Vitamin digestion and absorption e 2 50 3 443 0.0348134 ko00520 Amino sugar and nucleotide sugar metabolism e 2 50 3 443 0.0348134
Implementation
The GCNE is developed using C++ as open-source software under the GPLv3 license. In addition to our code, we used some third-party code in compliance with their licenses. The GCEN can be compiled and run in Linux, Windows, and macOS. Compiling the GCEN requires compiler and library support for the ISO C++ 2017 standard. The GCEN does not involve any new computational methods or algorithms but implemented and integrated some classic algorithms to annotate lncRNA more easily and faster. We described the main algorithms used as follows.
Data normalization
We have implemented four algorithms, including median normalization, quantile normalization [1], median-of-ratios method [2], and trimmed mean of M-values (TMM) [3]. For the median normalization, the median of gene expression values in each sample be calculated, then each gene expression value in the same sample multiply by the mean of all medians and divide by the median of this sample. The descriptions of the other three algorithms can be found in their original papers.
Co-expression network construction
We use the Spearman or Pearson correlation coefficients directly to determine co-expression patterns between gene pairs, which are more performing and robust [4]. The coefficient of Spearman or Pearson correlation ρ was calculated as follows.
For Pearson correlation, where or represents the vector of the expression value of each gene. or stands for each expression value, or , is the mean value of these expression values. For Spearman correlation, the vector of the expression value of each gene needs to be replaced by their ranks.
Module identification
The network modules, which are groups of genes with similar expression profiles, are explored based on the topological structure of the gene co-expression network [5]. Genes in the same module tend to be functionally related and co-regulated. We implemented a module identification algorithm based on the node similarity measure of their relative interconnectedness coupled with the hierarchical clustering method [6].
Where the is the similarity between gene and gene . The is the number of shared neighbor genes of gene and gene in the co-expression network. The is the adjacency of gene and gene . and are the connectivity of gene and gene , respectively.
Function annotation
After gene co-expression network construction and module identification, we use gene function enrichment to predict novel lncRNA’s or coding gene’s function. The value of enriched function was calculated as follows.
In this equation, is the total number of genes in the network. is the total number of genes having one certain annotation, is the number of a gene's immediate neighbors and is the number of neighbor genes having one certain annotation.
Another gene function analysis algorithm we implemented is the random walk with restart (RWR) [7], which measures each node’s relevance with respect to given seed nodes (here are genes with known function annotations) based on network propagation. The information (known gene function) is propagated through the edges from seed nodes to nearby nodes until convergence. To the end, prior information associating genes with a function of interest is super imposed on the nodes of the network.
Where represents our initial, or prior, information on genes. is a stochastic matrix, that is, its columns sum to 1. The parameter describes the trade-off between prior information and network smoothing. Repeated iteration of this equation converges to a steady-state. is the prior information associating genes with a function of interest. The implementation of RWR uses Eigen, which is a C++ template library for linear algebra (http://eigen.tuxfamily.org).
References
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK: limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015, 43(7):e47.
- Love MI, Huber W, Anders S: Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014, 15(12):550.
- Robinson MD, Oshlack A: A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 2010, 11(3):R25.
- Ballouz S, Verleyen W, Gillis J: Guidance for RNA-seq co-expression network construction and analysis: safety in numbers. Bioinformatics 2015, 31(13):2123-2130.
- Saelens W, Cannoodt R, Saeys Y: A comprehensive evaluation of module detection methods for gene expression data. Nat Commun 2018, 9(1):1090.
- Wang Z, San Lucas FA, Qiu P, Liu Y: Improving the sensitivity of sample clustering by leveraging gene co-expression networks in variable selection. BMC Bioinformatics 2014, 15:153.
- Cowen L, Ideker T, Raphael BJ, Sharan R: Network propagation: a universal amplifier of genetic associations. Nat Rev Genet 2017, 18(9):551-562.